De unde provine virusul SARS-CoV-2? Experții spun că provine de la lilieci, dar cum au ajuns la această concluzie?
Bioinformaticienii folosesc unelte precum BLAST, dar în acest articol vă vom arăta cum puteți analiza și voi, de acasă, genomul noului coronavirus din perspectiva științifică. Vom folosi metodele științifice pentru a prezice originea virusului SARS-CoV-2 comparând genomul său cu a altor coronavirusuri descoperite în alte specii de animale, precum rațe, bovine, găini și lilieci.
Sursa datelor
Secvența genomului SARS-CoV-2 este pusă la dispoziția publicului prin intermediul site-ului web al Bibliotecii Naționale a Medicinii din SUA: https://www.ncbi.nlm.nih.gov/labs/virus
 Observați două link-uri către două tipuri de secvențe de genom viral: nucleotide și proteine. Aici, vom analiza în mod special secvențele de nucleotide ale 4 specii de animale, și anume găini, lilieci, bovine și rațe și le vom compara cu secvențele de genom viral colectate de la persoanele infectate cu SARS-CoV-2.
Observați două link-uri către două tipuri de secvențe de genom viral: nucleotide și proteine. Aici, vom analiza în mod special secvențele de nucleotide ale 4 specii de animale, și anume găini, lilieci, bovine și rațe și le vom compara cu secvențele de genom viral colectate de la persoanele infectate cu SARS-CoV-2.
Obținerea datelor
Vom începe cu nucleotidele prin accesarea acestui link: https://www.ncbi.nlm.nih.gov
 Puteți observa că „Severe acute respiratory syndrome” este deja selectat în secțiunea „virusuri” din filtrul din partea stângă. Mai departe, sunt două tipuri de secvențe: „GenBank” și „RefSeq”. Vom alege „GenBank”, iar din secțiunea „Nucleotide Completeness” vom bifa „complete” și vom apăsa butonul „Download”. Bineînteles, puteți filtra rezultatele după multe alte criterii precum regiunea geografică, gazdă, data descoperirii, dar pentru acest experiment nu sunt necesare.
Puteți observa că „Severe acute respiratory syndrome” este deja selectat în secțiunea „virusuri” din filtrul din partea stângă. Mai departe, sunt două tipuri de secvențe: „GenBank” și „RefSeq”. Vom alege „GenBank”, iar din secțiunea „Nucleotide Completeness” vom bifa „complete” și vom apăsa butonul „Download”. Bineînteles, puteți filtra rezultatele după multe alte criterii precum regiunea geografică, gazdă, data descoperirii, dar pentru acest experiment nu sunt necesare.
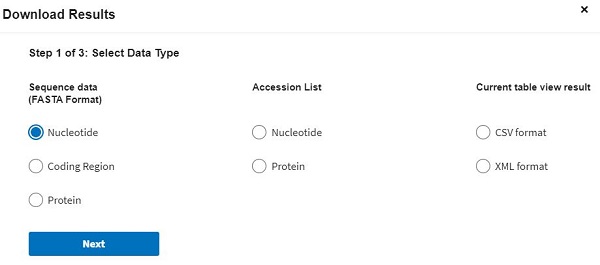
După ce apăsăm butonul „Download” va apărea o fereastră precum în imaginea alăturată de unde putem selecta tipul datelor, formatul de export etc. Vom selecta „Nucleotide”, apoi „Download all records” iar în pasul 3, „Use default”.
 După ce ați descărcat datele, puteți deschide fișierul în formatul „fasta” cu un editor de text precum Notepad, Notepad++ sau orice editor de text preferați. Fișierul va arăta precum în imaginea alăturată. Puteți observa că prima linie începe cu caracterul „>” care denotă descrierea virusului. În momentul scrierii acestui articol, există 3117 genomuri de virusuri complete (fișierul are aproximativ 92 MB).
După ce ați descărcat datele, puteți deschide fișierul în formatul „fasta” cu un editor de text precum Notepad, Notepad++ sau orice editor de text preferați. Fișierul va arăta precum în imaginea alăturată. Puteți observa că prima linie începe cu caracterul „>” care denotă descrierea virusului. În momentul scrierii acestui articol, există 3117 genomuri de virusuri complete (fișierul are aproximativ 92 MB).
Dacă doriți să săriți peste restul articolului, puteți încărca fișierul aici și vă va arăta secvențe similare, însă trebuie să reduceți dimensiunea (încărcând doar parțial fișierul) deoarece serverul are o limită a dimensiunii fișierului. Însă făcând acest lucru, pierdeți distracția. În continuare puteți analiza chiar voi genomurile virusurilor și să identificați care este sursa acestuia.
Acum, să descărcăm secvențele de nucleotide ale coronavirusurilor descoperite în alte specii de animale (rațe, găini, bovine și lilieci). Pentru a face acest lucru, trebuie să ștergem opțiunea din filtru numită „Severe acute respiratory syndrome coronavirus 2” și selectăm „Coronaviridae”. Acesta este numele științific pentru toate coronavirusurile care circulă în rândul tuturor speciilor. Apoi, din secțiunea „Host”, care înseamnă „gazdă”, selectăm „Gallus gallus” (găina) și apăsăm butonul Download. Repetăm pașii pentru celelalte 3 specii: bovine (Bos taurus), rațe (Anatidae) și lilieci (Chiroptera). Fișierele le-am denumit sequences_bovina.fasta, sequences_gaina.fasta, sequences_liliac.fasta. Nu are importanță denumirea, însă aceasta este folosită mai jos în cod.
Transformarea datelor
Acum că avem genomurile în 5 fișiere diferite (unul pentru noul coronavirus, SARS-CoV2 și 4 pentru cele 4 specii analizate ce conțin genomuri de coronavirusuri), primul pas este să scăpăm de liniile care încep cu „>”. Aceste linii nu ne interesează și pot afecta analiza pe care o vom efectua mai jos, deoarece avem nevoie doar de secvențele de nucleotide, nu și de meta-date. Bineînțeles, având milioane de linii într-un fișier, nu le vom șterge manual, ci vom folosi un mic script. Pentru a rula codul din secțiunile următoare veți avea nevoie de Jupyter, un mediu de dezvoltare și câteva librarii: ntlk, care este o librărie pentru procesarea limbajului natural (uman), sklearn, care este un framework pentru analiza datelor și matplotlib, folosit pentru vizualizări grafice (opțional).
Bineînteles, puteți folosi orice alte unelte de transformare, extragerea caracteristicilor și „tokenizare”. Dacă sunteți familiarizați cu C# / .NET, atunci o variantă bună este StandfordNLP.
Pentru a identifica fiecare nucleotidă (folosind semnul „>”) se va rula codul de mai jos:
from nltk.corpus import stopwords
from sklearn.feature_extraction.text import CountVectorizer
import pandas as pd
def process_file(filename,target_val):
f = open(filename) #’datafolder\\sequences_gaina.fasta’)
lines = „”
s1 = list()
step = 0
term = 0
for line in f:
line = ”.join(line.split())
if line.startswith(„>”) and step==0:
line = line.split(‘>’,1)[0].strip()
step = step + 1
if line.startswith(„>”) and step>=1: #and step>=1:
line = line.split(‘>’,1)[0].strip()
s1.append(lines)
lines = „”
step = step + 1
term = 0
lines = lines + line
Reprezentarea numerică a secvențelor
Următorul pas este de a genera caracteristici folosind șirurile care conțin caracterele noastre de secvență. Deoarece secvența este reprezentată ca o serie de caractere alfabetice, trebuie să împărțim secvența în bucăți mai mici, numite token-uri – de exemplu: un token cu 2 caractere, unul cu 3, altul doar cu 4 etc. Apoi, trebuie să transformăm token-urile într-o reprezentare numerică, astfel încât să le putem introduce în algoritmul de învățare folosit ulterior pentru predicție. Pentru a face acest lucru, folosim conceptul de reprezentare numit „bag-of-words” care calculează frecvența, sau de câte ori apare, a fiecărui token. Astfel obținem două lucruri importante: vocabularul (token-urile) și numărul de apariții pentru fiecare token.
Pentru a aplica în același timp ambele metode, n-grams și bag-of-words, se poate folosi metoda CountVectorizer din librăria scikit-learn. Metoda primește 3 parametri, definiți ca în codul alăturat.
count_vect = CountVectorizer(lowercase=False, ngram_range=(2,4),analyzer=’char’)
X1 = count_vect.fit_transform(s1)
Pentru această etapă, vom seta mărimea token-urilor la 2 până la 4 caractere. Astfel ne va genera token-uri precum „AT” (2 caractere), „ACG” (3 caractere) și „ACTT” (4 caractere).
def generate_ngrams(s1):
count_vect = CountVectorizer(lowercase=False, ngram_range=(2,4),analyzer=’char’)
X1 = count_vect.fit_transform(s1)
lcount = list()
lcount = []
for i in s1:
count = len(i)
#print(count)
lcount.append(count)
count_vect_df = pd.DataFrame(X1.todense(), columns=count_vect.get_feature_names())
count_vect_df=count_vect_df.apply(lambda x: x / lcount[x.name] ,axis=1)
return count_vect_df
Etichetarea datelor
O dată ce datele au fost transformate în valori numerice, trebuie să le punem o etichetă pentru predicție.
df1 = process_file(‘datafolder\\sequences_gaina.fasta’,”gaina”)
df2 = process_file(‘datafolder\\sequences_rata.fasta’,”rata”)
df3 = process_file(‘datafolder\\sequences_bovina.fasta’,”bovina”)
df4 = process_file(‘datafolder\\sequences_liliac.fasta’,”liliac”)
Puteți observa că fiecare fișier fasta este atribuit cu propria sa etichetă (găină, rață, bovină și liliac). Bineînteles, modificați calea către fișier și denumirea acestuia dacă le-ați salvat cu un alt nume.
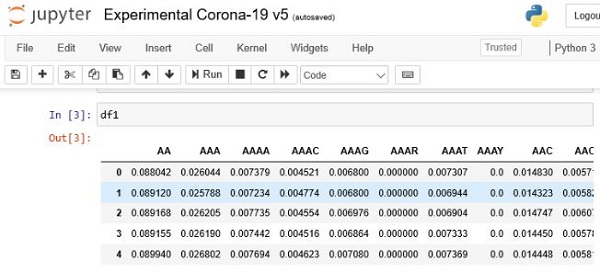
Dacă totul a decurs fără erori, ar trebui să vedeți un rezultat precum cel de mai jos, unde toate caracteristicile au fost transformate în valori numerice.
 Analiză exploratorie
Analiză exploratorie
Având toate secvențele reprezentate, putem explora distribuția procentuală a seturilor de date pentru a vedea în rândul căror specii aceste virusuri sunt mai răspândite.
Construirea modelului predictiv
Acum suntem pregătiți pentru a construi modelul predictiv trimițând seturile de nucleotide către algoritmul de „machine learning”. În acest caz, este folosit algoritmul „gradient boosting” pentru modelul predictiv.
from sklearn.model_selection import train_test_split
from xgboost import XGBClassifier
from xgboost import plot_importance
import xgboost
# create a train/test split
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=7, shuffle=True)
model = XGBClassifier()
model.fit(X_train, y_train)
Folosind modelul, se poate analiza mai departe pentru a determina ce secvențe de nucleotide sunt mai răspândite și au un factor determinant pentru a identifica gazda virusului.
Transformarea datelor și reprezentarea
Mai departe, trebuie să încărcăm fișierul care conține noul coronavirus, SARS-CoV-2 (primul fișier descărcat) în aplicație pentru a efectua predicția.
cov = process_file(‘datafolder\\serquences.fasta’,“SARS-CoV-2”)
cov
Următorul pas este antrenarea algoritmului prin compararea rezultatelor și renunțând la prima linie deoarece nu avem nevoie de etichetă în acest moment:
cov = cov.drop(‘target’, axis=1)
mc = X_train.columns.difference(cov.columns)
mc
Putem observa că anumite caracteristici precum „AAAM”, „AAR”, „AAAY” sunt găsite în coronavirusuri dar nu și în SARS-CoV-2.
În următorul pas, scoatem caracteristicile care există în SARS-CoV-2 dar nu există în celelalte fișiere. O data ce am găsit care sunt acestea, le ștergem:
rf = cov.columns.difference(X_train.columns)
cov = cov.drop(rf, axis=1)
Rezultatul
Acum suntem pregătiți să prezicem originea virusului SARS-CoV-2! Tot ce trebuie să faceți este să rulați asta:
model.predict(cov)
iar rezultatul:
array([‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’,
‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’,
‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’,
‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’, ‘liliac’], dtype=object)
Pe baza valorilor prezise, putem observa că toate sample-urile rămase au prezis că originea virusului este liliacul. Nici măcar una nu a prezis alt animal. Mai departe, se poate folosi „predict_proba” pentru a obține probabilitatea pentru fiecare predicție. Rezultatul a fost de 0,957, adică 95,7% șanse ca originea virusului să fie de la lilieci, în timp de doar 4,1% șanse de la rațe și 0,1 de la găină sau bovine.
Compararea rezultatelor cu BLAST
BLAST este metoda standard de a compara secvențele de nucleotide sau proteine. Programul se poate găsi aici: https://blast.ncbi.nlm.nih.gov/Blast.cgi unde puteți încărca fișierul conținând secvențele de SARS-CoV-2 și vă va arăta secvențe similare. Însă spre deosebire de metoda din acest articol, BLAST nu aplică modele predictive ci folosește un algoritm de căutare euristic.
Bibliografie:
https://towardsdatascience.com
Link-uri și aplicații utile:
Word2Vec
Jupyter
Matplotlyb
Standford Natural Language Processing
NTLK









{kind=link}